 |
since 2003.4.15
This paper was presented in US-Japan Binational Workshop on Molecular Evolution held at the Graduate University for Advanced Studies, Hayama, Japan, August 25-27, 1995, and printed in "Current Topics on Molecular Evolution" 9-18. 1995.
| |
To clarify the origin and phylogeny of Hawaiian drosophilid species, we determined Adh sequences for 14 species of the genus Drosophila and related genera. Together with homologous sequences published for 39 species, a phylogenetic tree was constructed and its statistical reliability was tested. Using the Adh sequences from clear-cut outgroups, we confirmed that the subgenus Scaptodrosophila is the outgroup of the rest of the genus Drosophila and of its related genera and that Hawaiian drosophilids belong to the subgenus Drosophila of the genus Drosophila. The phylogenetic tree obtained showed that Hawaiian drosophilids are more closely related to the virilis-repleta lineage than to the immigrans-Hirtodrosophila lineage. Both Hawaiian and non-Hawaiian species of the genus Scaptomyza and the species of the subgenus Engiscaptomyza form a monophyletic cluster as the closest sister group of the Hawaiian Drosophila, supporting Throckmorton's hypothesis for the origin of the genus Scaptomyza in Hawaii. Additionally, it was found that the second intron is missing in the D. sexvittata (the subgenus Hirtodrosophila) Adh gene. This qualitative information is in accordance with the phylogenetic position obtained for Hawaiian drosophilids being not closely related to Hirtodrosophila.
The Hawaiian islands preserve one of the richest endemic drosophilid faunas on earth due to their suitable climate and good geographic isolation from continents. Furthermore, these islands provide chronological information, which is of importance for evolutionary study. During the last million years, the earth's volcanic activity has extruded lava periodically from the deep mantle at a fixed "hot spot" in the middle of Pacific ocean. These volcanic eruptions have built up islands above the sea, and the tectonic movements of the Pacific Plate have continuously carried them northwest. Consequently, ages within the chain of the current Hawaiian islands range from 5-6 million years for Kauai on the northwest end to 700,000 years for Hawaii on the southeast end. This chain of islands continues northwestward further to the island of Midway (27 million years old) and even to Emperor Seamounts (55-70 million years old) (see Hardy and Kaneshiro 1981 for review). In this context, the evolution of Hawaiian drosophilids has been thoroughly studied from various points of view, including morphology (Throckmorton 1966, Kaneshiro 1969), behavior (e. g., Kaneshiro 1980), cytology (Yoon et al. 1972, 1975), and molecular phylogenetics (Beverley and Wilson 1985; DeSalle 1992; Thomas and Hunt 1991, 1993; Russo et al. 1995). In spite of these studies, the origin of Hawaiian drosophilids or the phylogenetic relationship to other continental drosophilids is still controversial.
 |
 |
Therefore, to determine the phylogenetic relationships among the species groups of the genus Drosophila and its related genera, we analyzed nucleotide sequences of the alcohol dehydrogenase (Adh) gene. Similar phylogenetic studies for the drosophilid Adh gene have been already made by Thomas and Hunt (1993) and Russ et al. (1995). Both studies showed high statistical reliability, suggesting the appropriateness of the Adh gene for phylogenetic study of drosophilids. However, since the species examined were confined to a small number of species groups, the origin of Hawaiian drosophilids was not resolved. Therefore, to determine the origin of Hawaiian drosophilids, we determined the Adh sequences for 14 species from various species groups and related genera. Together with homologous sequences published for 39 species, we reconstructed a phylogenetic tree for 53 species belonging to the family Drosophilidae and discussed the origin of Hawaiian drosophilids based on the molecular phylogeny.
DNA Cloning and Sequencing
The species
for which the Adh sequence was analyzed are listed in table 1. The Adh
sequences were obtained by PCR amplification using mRNA or genomic DNA,
extracted from adult flies according to Chomczynski and Sacchi's (1987)
or Steller's (1990) method, as the templates. The primers used were 5'-AACAAGAA(TC)(AG)T(AGTC)(AG)T(AGTC)TT(TC)GT-3'
and 5'-TAGAT(AGTC)(TC)(GC) (AGTC)GA(AG)TCCCA(AG)TG-3', where the nucleotides
in the parentheses indicate degeneracy. The amplified DNA fragments were
cloned into plasmid vector pUC118 by using E. coli K12.MV1184 as
a host. The recombinants were then subcloned by using exonucleaseIII and
mungbean nuclease (Henikoff 1984). Single strand templates for DNA sequencing
were then obtained from these subclones by taking advantage of the pUC118/119-M13KO7
system (Vieira and Messing 1987). The nucleotide sequences were determined
by using an ABI autosequencer according to the protocol supplied by the
manufacture.
 |
DNA Sequence Data
The Adh sequences for other 39 species were almost the same with those
analyzed by Russo et al. (1995). While Russo et al. (1995) used both of
the duplicated genes, i. e., Adh-1 and Adh-2, for several species, we used
only Adh-2. This is because, first, we obtained the Adh gene of many species
from adult mRNA that was most probably transcribed from Adh-2 even if the
Adh gene was duplicated and, second, selections from the duplicated genes
seemed not to be important for estimating phylogenetic relationships between
less closely related species, according to Russo et al.'s (1995) study.
As a result, we did not include the sequence for D. navojoa since
only the sequence of Adh-1 was available, whereas we included the sequence
for D. erecta which was published very recently.
Phylogenetic Analyses
The protein-coding regions between the two primer sites were used for
phylogenetic analysis. Thus, the number of nucleotide sites examined was
711. The phylogenetic tree was constructed by the minimum evolution (ME)
method (Rzhetsky and Nei 1992) with Jukes and Cantor's (1969) distance
for estimating the number of nucleotide substitutions between sequences.
The ME tree obtained was then statistically evaluated by the confidence
probabilities for each interior branch. For the estimation and the statistical
tests of ME trees, we used the METREE program kindly provided by A. Rzhetsky.
 |
Phylogenetic relationships for drosophilids inferred from the Adh
gene
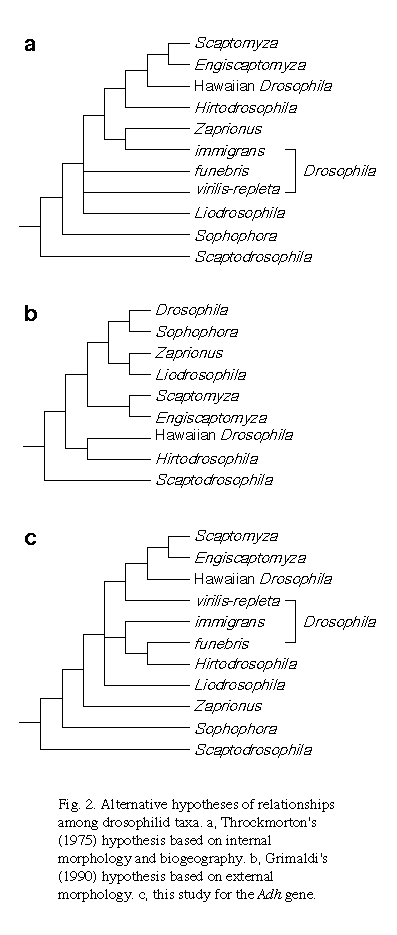 |
The origin of Hawaiian drosophilids
It has been believed by many drosophilists that Hawaiian drosophilids
originated in the immigrans-Hirtodrosophila radiation and therefore
should be closely related to the species groups including D. histrio,
D. confusa, D. deticeps, and so forth (Throckmorton 1975;
see also Okada 1967, 1971, and Hardy and Kaneshiro 1981). Since Adh sequences
for these species were not available, we determined the Adh sequences from
D. sexvittata, D. brachynephros, and D. albomicans,
which belong to Hirtodrosophila, the quinaria group, and
the immigrans group, respectively, and included them in the phylogenetic
analysis. In our tree, however, the close relationship between Hawaiian
drosophilids and the immigrans-Hirtodrosophila lineage can not be
found. Instead, the cluster of Hawaiian drosophilids forms a tight cluster
with that of the virilis-repleta lineage (CP = 99%), suggesting
that Hawaiian drosophilids diverged from the virilis-repleta lineage
after its divergence from the immigrans-Hirtodrosophila lineage.
Another unsolved problem concerning the origin of Hawaiian drosophilids
is the origin of the genus Scaptomyza. It seems to have been a consensus
to date that Hawaiian drosophilids are classified into two major taxa,
"drosophiloid" and "scaptomizoid" as designated by
Throckmorton (1975). The former mostly belongs to the genus Drosophila
and the latter mostly belongs to the genus Scaptomyza. Since
morphological distinctions between the two genera tend to disappear in
Hawaiian drosophilids, Throckmorton (1966) hypothesized that the genus
Scaptomyza originated in Hawaii and subsequently dispersed to the
rest of the world. Alternatively, it is also possible that the ancestors
of the drosophiloid and the scaptomizoid lineages came into Hawaii separately.
To examine these alternative hypotheses, we determined and included into
our analysis the Adh sequences from non-Hawaiian Scaptomyza species,
S. graminum of the subgenus Scaptomyza and S. pallida
of the subgenus Parascaptomyza. The interpretation of the results
is rather complicated. While all the Scaptomyza species and Engiscaptomyza
(D. crassifemur) form a tight monophyletic cluster (CP = 99%),
the relationships between Hawaiian and non-Hawaiian species cannot be resolved.
It is apparent, however, that Engiscaptomyza is a member of the
genus Scaptomyza according to our tree.
Loss of an intron
The basic structure of the Adh gene is known to be organized into four
exons divided by three introns (see e. g., Kreitman 1983). However, it
was found that in the D. sexvittata Adh gene the second intron is
missing and the second and third exons are fused into a single exon. The
same loss of the second intron has been suggested in the Adh gene of Mycodrosophila
biceps because of the shorter DNA fragments amplified by PCR (data
not shown). While a similar phenomenon was found in the D. willistoni
Adh gene (Anderson et al. 1993), in which the third intron is missing,
this is the first report of the lack of the second intron in the Drosophila
Adh gene. Since no species other than those related to Hirtodrosophila
are known to have the Adh gene without the second intron, this feature
may be a good criterion for the identification of the Hirtodrosophila
lineage. Since the second intron of the Adh gene is present in Hawaiian
drosophilids, it is obvious that Hawaiian drosophilids did not originate
from Hirtodrosophila at least after the loss of the intron.
In this study, we have produced a phylogenetic tree for the Adh gene from a wider variety of drosophilid species than previous studies by Thomas and Hunt (1993) and Russo et al. (1995). Despite the larger number of species used, our tree is statistically well supported as the previous trees for a smaller number of species are. To infer the origin and phylogenetic relationships of Hawaiian drosophilids, we especially focus on resolving which species group is most closely related to Hawaiian drosophilids and on whether Hawaiian and non-Hawaiian Scaptomyza form a monophyletic cluster. Most interestingly, our analysis placed the cluster of Hawaiian drosophilids on the virilis-repleta lineage rather than on the immigrans-Hirtodrosophila lineage. Although the close relationship between Hawaiian drosophilids and the virilis-repleta lineage was also suggested by comparisons of morphological characters for male paragonia (Throckmorton 1966) and of banding patterns in a chromosome segment (Stalker 1972), many drosophilists have believed that Hawaiian drosophilids are closely related to the immigrans-Hirtodrosophila lineage. This conjecture is based mainly on similarities in internal morphology. For example, both taxa share the characteristic feature of ventral receptacles with proximal coils and distal folds (Okada 1967). The present result is apparently in contradiction to this traditional view. A parsimonious interpretation of the contradiction may be that the feature of the ventral receptacles is ancestral and shared by the common ancestor of the two lineages. Usually, folded forms are characteristic of the subgenus Sophophora, whereas coiled forms are characteristic of the subgenus Drosophila. However, since Leucophenga, Scaptodrosophila, and Zaprionus also possess the folded forms, assuming that the folded forms are ancestral is not difficult. If it is true, then the transition from the folded to the coiled forms must have occurred in the common ancestor of the subgenus Drosophila, and both Hawaiian drosophilids and Hirtodrosophila may have retained the transient forms. Similarly, both forms of the ventral receptacles are observed in Liodrosophila: a coiled form in Li. aerea and a folded form in Li. bicolor (Okada 1956). This is consistent with the interpretation, since Liodrosophila is placed between Zaprionus and the subgenus Drosophila in our tree. Alternatively, it is also possible that the forms of the ventral receptacles occurred convergently in the different lineages.
 |
Another feature supporting a close relationship between Hawaiian drosophilids and Hirtodrosophila is the fact that both the Hawaiian Drosophila and some species of Hirtodrosophila have morphological features somewhat intermediate between those of the genera Drosophila and Scaptomyza (Okada 1971). As in the case of the internal morphology mentioned above, a parsimonious interpretation may be that some ancestral species of the subgenus Drosophila had the intermediate features. Before we come to this conclusion, however, we should analyze Adh sequences from those species like D. denticeps and species of Lordiphosa, which are known to have intermediate characteristics. The phylogenetic position of these species is especially interesting, since in this study we could not determine a particular species or species group which takes the position of the closest sister group of Hawaiian drosophilids. The best candidates would be species showing intermediate characteristics. Similarly, Adh sequences from many species or species groups in the virilis-repleta lineage should be examined as well.
It is apparent from this study that Scaptomyza (including Engiscaptomyza) forms a monophyletic cluster. We included species from the subgenera Scaptomyza and Bunostoma, both of which were thought to be located in the outermost position of the Scaptomyza cluster (see Grimaldi 1990 for review). Therefore, it is unlikely that the observed monophyly is due to the small number of species examined. It is also apparent that the Scaptomyza cluster takes the position as the closest sister group of the drosophiloid (the Hawaiian Drosophila). These results support Throckmorton's (1966) hypothesis that both the drosophiloids and scaptomyzoids originated from the same ancestor and that Scaptomyza subsequently dispersed to the rest of the world. Although the alternative hypothesis that the ancestors of the drosophiloids and of the scaptomyzoids were independently introduced into Hawaii twice may be equally parsimonious in terms of the number of migratory events, we prefer the former alternative for the following reasons. First, as discussed by Throckmorton (1966), the latter alternative entails postulating the unrealistic coincidence that the two different ancestors were so closely related. Second, the extent of the largest sequence divergences within Hawaiian Scaptomyza species is the same as that within non-Hawaiian species in our tree. This is also unlikely under the latter hypothesis. As discussed earlier, it is unlikely that the extent of sequence divergences is underestimated due to a small number of species examined. To test the alternative hypotheses, it will be necessary to determine further Adh sequences from related taxa such as Lordiphosa and Titanochaeta.
Assuming the divergence time of 11 million years between S. albovittata and D. crassifemur (Thomas and Hunt 1993; Russo et al. 1995), the divergence time for the deepest root of Scaptomyza is estimated to be 14 Mya, based on the average distance between the S. albovittata/pallida/D. crassifemur and S. graminum/anomala/palmae clusters. This divergence time conflicts with the time (»23 Mya) estimated for the Scaptomyza species found in Dominican amber (Grimaldi 1987), if the origin of Scaptomyza in Hawaii is correct. However, we are skeptical about that the fossil Scaptomyza species really belongs to the extant genus Scaptomyza. Actually, the fossil species possesses none of the diagnostic morphological features of Central American species of Scaptomyza (Grimaldi 1987). If we assume that some ancestral species of the subgenus Drosophila had morphological features found in the present Scaptomyza mentioned above, this conflict may be explained. The morphological features of the ancestral species remain unsolved, awaiting further studies. As stated by many authors, drosophilids are an excellent material for studying mechanisms of evolution. Therefore, it is important to establish their phylogenetic relationships as well as to study their biology, which is still poorly known except for particular species.
We would like to thank Claudia Russo for giving us their sequence data set and Marcy Uyenoyama and Masatoshi Nei for their suggestions for improving the manuscript. Special thanks to Hideaki Watabe due to his assistance in species identification for flies collected from fields. This work was supported by grant number 06640906 from the Ministry of Education, Science and Culture of Japan to T. A.